Time:2021-12-22
A recent study published in Movement Disorders demonstrated that the truncated spastin may damage the corticospinal tracts in hereditary spastic paraplegia (HSP) patients via an isoform-specific toxic effect. This work was performed by Dr. LIU Jing-Yu’s lab at the Institute of Neuroscience (ION), CAS Center for Excellence in Brain Science and Intelligence Technology of the Chinese Academy of Sciences.
The LIU’s lab successfully identified the pathogenic gene SPAST in three hereditary spastic paraplegia (HSP, SPG4 subtype) families by linkage analysis and Sanger sequencing. Further functional analyses of the mutant proteins by Western blotting and immunofluorescence showed that the truncated SPASTIN encoded by the mutant SPAST can disturb microtubule dynamics through the isoform-specific effects.
The team further proposes that truncated forms of SPASTIN may affect neuronal function in patients through their long-term cellular accumulation, providing new directions for exploring the pathogenic mechanisms and treatments of SPG4 subtype.
HSP is a genetically and clinically heterogeneous neurodegenerative disorder mainly characterized by progressive spasticity and weakness in the lower extremities. Haploinsufficiency is generally proposed as the mechanism underlying HSP. But the correlation between the clinical symptoms and the degree of impaired protein function has not been well elucidated, and advances in clinical treatments has been very slow.
In this study, the LIU’s team investigated three large families with pure HSP (247 members, 67 patients, Figure 1) and identified a novel mutation (c.985dupA/p.Met329Asnfs*3) in SPAST by linkage analysis and Sanger sequencing. The mutation did not promote mRNA decay but led to the production of two truncated mutants (mutant M1 and M87 isoforms), which were found to be more stable than the corresponding wild-type isoforms. Furthermore, they showed that the mutant M1 isoform heavily decorated the microtubules and rendered them resistant to depolymerization. In contrast, the mutant M87 isoform was diffusely localized in both the nucleus and the cytoplasm, could not decorate the microtubules, and was not able to attenuate microtubule disassembly (Figure 2A).
In this study, the LIU’s team demonstrated that the truncated SAPSTIN can accumulate intracellularly over long-term periods, thereby potentially resulting in cytotoxicity in the corticospinal tract with high expression levels of SAPSTIN. Meanwhile, they further confirmed that mutant SPASTIN may lead to HSP by affecting the corticospinal tract and based on current evidence, this effect is isoform-specific (Figure 2B). More studies on the exact mechanisms of its neural toxicity are being carried out.
CHEN Rui, DU Shiyue (College of Life Science and Technology, Huazhong University of Science and Technology) and YAO Yanyi (Medical Genetics Center, Maternal and Child Health Hospital of Hubei Province), who were supervised by Dr. LIU Jing Yu and postdoctoral scholar XU Xuan, contributed equally to this work. This work was supported by the National Natural Science Foundation of China, Ministry of Science and Technology of the People′s Republic of China,, and Shanghai Municipal Government .
https://doi.org/10.1002/mds.28885
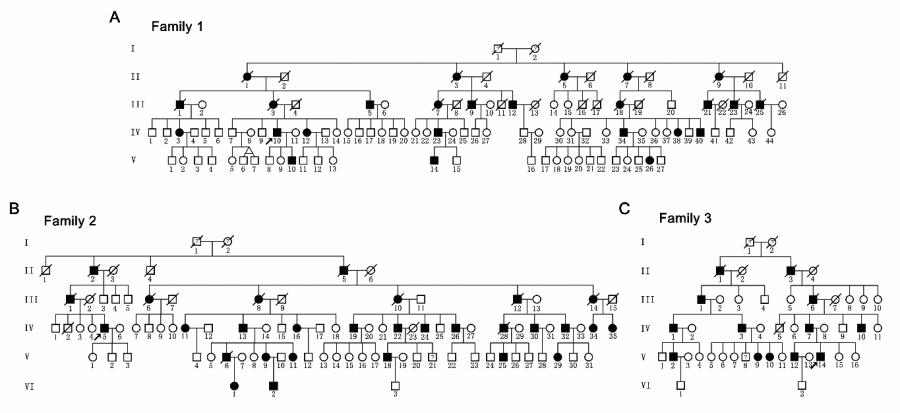
Figure 1. Pedigrees of three hereditary spastic paraparesis-affected families with autosomal dominant Mendelian inheritance.
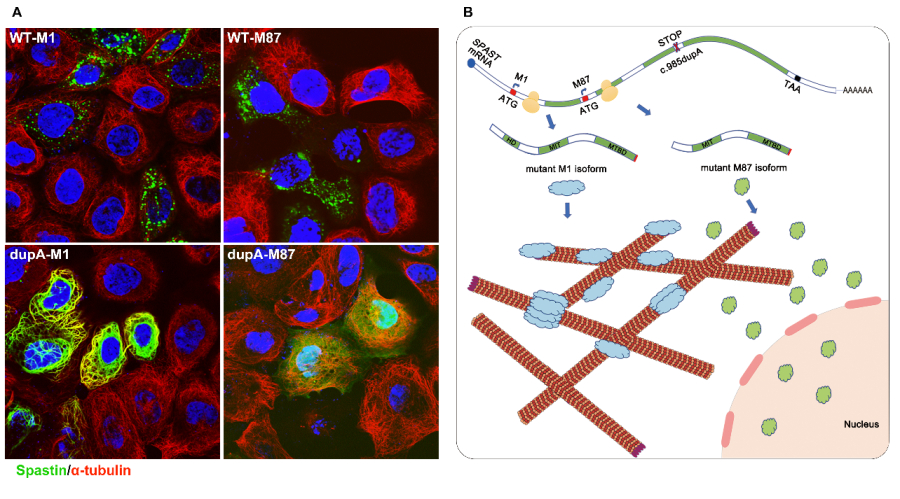
Figure 2. (A) Subcellular localization of wild type (WT-M1 and WT-M87) and truncated (dupA-M1 and dupA-M87) spastin. (B) Hypothetical pathogenic mechanism for mutant c.985dupA (p.Met329Asnfs *3).
 附件下载:
附件下载: 
