Time:2019-06-03
A recent study published in PNAS implicates a critical role of UBE3A-PTPA-PP2A signaling in the pathogenesis of UBE3A-related disorders and suggests a target for therapeutic intervention to Angelman syndrome and autism. This work was performed by researchers in Dr. XIONG Zhiqi’s Lab at the Center for Excellence in Brain Science and Intelligence Technology, Institute of Neuroscience, State Key Laboratory of Neuroscience, Chinese Academy of Science and Prof. LIAO Lujian’s Lab at East China Normal University.
The imprinted gene UBE3A encodes a HECT (homologous to E6 associated protein carboxy terminus) domain E3 ligase UBE3A, which conjugates poly-ubiquitin chains to specific lysine residues in its substrates, regulating the expression and function of these proteins. Deletion or loss-of-function mutations of the maternally inherited allele of UBE3A result in Angelman syndrome (AS), a neurodevelopmental disorder characterized by severe developmental delay, intellectual disability, motor dysfunction and seizures. On the other hand, maternal duplication or triplication of the chromosome 15q11-13 region, where UBE3A resides, is associated with autism. These findings demonstrate that UBE3A plays a critical role in brain development and function. However, the potential mechanism underlying its dysfunction in neurodevelopmental disorders is not well understood. Previous studies have reported few UBE3A targets in the brain. Unfortunately, these substrates cannot yet account for the brain-related symptoms reported in UBE3A-related disorders.
In this study, researchers found that the activity of protein phosphatase 2A (PP2A) was upregulated in the AS model mice (Ube3am-/p+) at early developmental stages. The mRNA and protein levels of PP2A subunits were unchanged in Ube3am-/p+ mice, although the activity of PP2A was regulated by UBE3A. To investigate why PP2A activity increased in AS mice, they systematically screened the alterations of proteins in AS model mice using stable isotope labeling of amino acids in mammals (SILAM) combined with quantitative mass spectrometry. Interestingly, the activator of PP2A, PTPA, was elevated in Ube3am-/p+ mice. Furthermore, using in vivo and in vitro ubiquitination assays, they demonstrate that PTPA is a bona fide ubiquitin substrate of UBE3A. PTPA is known to regulate the phosphorylation and/or methylation of the catalytic subunit PP2Ac to promote PP2A holoenzyme assembly in nonneuronal cells. Using biochemical methods, this study demonstrates that UBE3A specifically regulates the methylation of PP2Ac and promotes the assembly of PP2A holoenzymes including the B55 family.
Overexpression of PTPA in cultured neurons caused phenotypes similar to those observed in Ube3am-/p+ mice. Increased PTPA led to the reduction of the proportion of mature spines, with a concomitant increase in immature spines. Genetical reduction of PTPA reversed this spine defects in Ube3am-/p+ mice. Remarkably, inhibition of PP2A activity with a small molecule LB-100 could significantly ameliorate cellular and behavioral defects in UBE3A deficient mice, including synaptic transmission, spine morphology, and motor skills. LB-100 has recently been tested in a phase I clinical trial for treatment of cancer and found to be safe.
Together, this study offers a novel understanding of the molecular mechanism underlying the pathology of UBE3A-related neurodevelopmental disorders and suggests that PP2A-based drugs could be potential therapeutic candidates for the treatment of these disorders.
This work entitled “UBE3A-mediated PTPA ubiquitination and degradation regulate PP2A activity and dendritic spine morphology” was published in PNAS on June 3. This work was carried out by WANG Jie and his colleagues. This work was supported by grants from the Chinese Academy of Sciences (XDB32060000, QYZDJ-SSW-SMC010), the Ministry of Science and Technology (2016YFA0501002), and the Science and Technology Commission of Shanghai Municipality (16JC1420202).
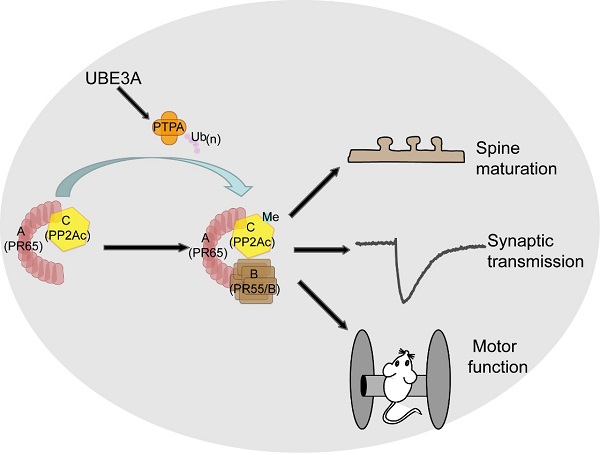
Figure. A model of UBE3A-PTPA-PP2A signaling.
 附件下载:
附件下载: 
