Time:2018-09-27
Ion channels are pore-forming membrane proteins that selectively allow ions to flow across the cell membrane; they are the molecular basis for the cell excitability. Malfunction of ion channels causes over 60 different inherited human diseases known as channelopathies. Inherited mutations result in dysfunctions of ion channels via different mechanisms: abnormal transcription/translation, deficient assembly or protein trafficking, defective channel gating/kinetics, and altered channel permeability. Different disease-related mutant channels thus may need different strategies to rescue their function. Current drug screening mainly focuses on identification of compounds that modulate the gating and kinetics of ion channels. However, the cumulative evidence suggests that defective protein biogenesis represents a rather common mechanism underlying many channelopathies, including LQTS, cystic fibrosis, episodic ataxia type 1, epilepsy, and hearing loss. There is an urgent need to develop different screening methods for the discovery of drugs that enhance or suppress specific mechanisms causing abnormal channel phenotypes underlying channelopathies.
A recent study published in the Nature communications reported an in vivo high-throughput method for screening compounds that have therapeutic potential in treating channelopathies. This work was performed by Dr. CAI Shiqing’s lab at the Institute of Neuroscience, State Key Laboratory of Neuroscience, Center for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Science and Dr. LAN Feng’s team at Beijing Anzhen Hospital, Capital Medical University.
In the current study, the authors have developed a novel in vivo screening tool for the discovery of drugs targeting the hERG channel, a voltage-gated K+ channel that mediates cardiac IKr currents. Dysfunction of hERG K+ channels due to genetic mutations or drug-induced inhibition results in Long QT syndrome (LQTS), a life-threatening cardiac arrhythmia. The channel shows homology with the C. elegans ERG-type K+ channel UNC-103, which regulates worms' locomotion, egg-laying and male mating behaviors. The authors generate transgenic animals by expressing hERG K+ channels (or its disease-related mutants) in C. elegans. The transgenic worms show severe egg-laying and locomotive defects, which offer indicators for screening small-molecule channel modulators. Phenotype-based screens in these transgenic worms identify alphitolic acid (ALA) as a novel hERG trafficking inhibitor and Prostratin and ingenol 3,20-dibenzoate (IDB), as functional correctors of some hERG mutant channels. Importanly, prostratin and IDB correct the electrophysiological abnormality of cardiomyocytes carrying a trafficking-defective LQTS2 mutation A561V. These compounds promote hERGA561V membrane trafficking by activating PKCε signaling and consequently phosphorylating the S606 site at the pore region of the channel. Thus, this study has demonstrated that mutation-specific screening could be achieved by using C. elegans animal models of channelopathies. The study will definitely help to develop precise medicine approach to treating channelopathies.
This work was mainly conducted by graduate student JIANG Qiang and LI Kai under the supervision of Dr. CAI Shiqing and LAN Feng. LU Wen-Jing, LI Shuang, CHEN Xin, LIU Xi-Juan and YUAN Jie also contributed to this work. This work was supported by the Strategic Priority Research Program of Chinese Academy of Science, Natural Science Foundation of China and the National Key R&D Program of China.
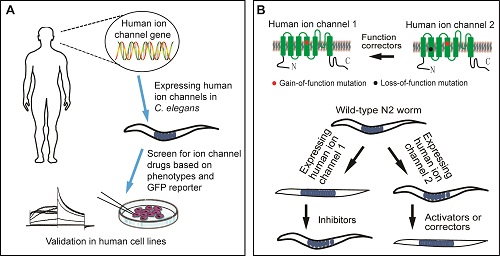
Figure legends: Schematic illumination of generating animal models of channelopathies and strategy for drug screening.
 附件下载:
附件下载: 
