Time:2018-07-24
A recent study published in the Journal of Neuroscience described an important mechanism that helps to understand the effects of the C9orf72 hexanucleotide repeat expansion in synaptic regulation and excitotoxicity, and in the development of C9orf72-related amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). This study was performed by Dr. XU Wangchao under the supervision of Dr. XU Jin at the Laboratory of Mechanisms of Neurodegenerative Diseases, Institute of Neuroscience, State Key Laboratory of Neuroscience, Chinese Academy of Sciences.
ALS is a devastating motor neuron disease characterized by fast progression and debilitating effects. Recent studies have indicated that ALS is genetically and clinically associated with FTD, the second common familial form of dementia. Some patients exhibit features of both diseases. Furthermore, mutations in a few genes could cause both ALS and FTD. The most common cause of familial ALS and FTD is the unusual (GGGGCC)n hexanucleotide repeat expansion mutation in C9orf72. However, the mechanism whereby the repeat expansion causing ALS and FTD is unclear.
In this study, Drs. XU Wangchao and XU Jin took advantage of powerful genetic tools in Drosophila model and investigated the neurotoxic mechanism of the arginine-rich dipeptide repeats (DPRs), which are protein products derived from the C9orf72 hexanucleotide repeat expansion. By expressing DPRs with different toxicity strength in various neuronal populations in a Drosophila model, they unexpectedly found that GR/PR with 36 repeats could lead to neurodegenerative phenotypes only when they were expressed in glutamatergic neurons, including motor neurons. They further detected increased extracellular glutamate and intracellular calcium levels in GR/PR-expressing larval ventral nerve cord and/or adult brain, accompanied by the significant increase of synaptic structure in larval neuromuscular junctions. Inhibiting the vesicular glutamate transporter (vGlut) expression or blocking the NMDA receptor in presynaptic glutamatergic motor neurons could effectively rescue the motor deficits and shortened life span caused by poly GR/PR, thus indicating a cell-autonomous excitotoxicity mechanism. This mechanism is vastly different from the commonly recognized non-cell autonomous mechanism involving glia and motor neurons in ALS, and it could also apply to glutamatergic neurons that are mainly affected in FTD. Therefore, this new study has revealed a novel mode of synaptic regulation by products derived from C9orf72 hexanucleotide repeat expansion and provided evidence to support the use of NMDA receptor antagonists in the treatment of C9-related ALS and FTD.
This work entitled “C9orf72 dipeptide repeats cause selective neurodegeneration and cell-autonomous excitotoxicity in Drosophila glutamatergic neurons” was published online in the Journal of Neuroscience On July 24 . This study was funded by grants from National Science Foundation of China and Chinese Ministry of Science and Technology.
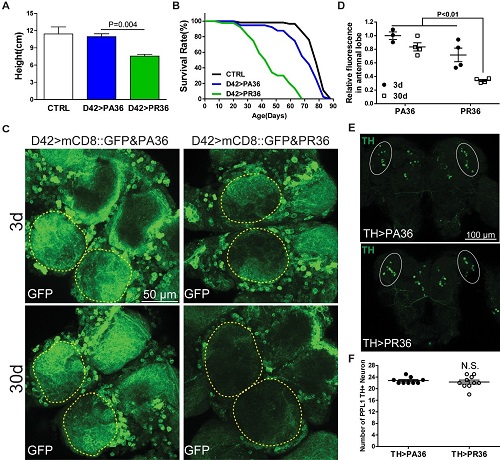
Fig.1. Representative images showing the loss of motor neurons expressing PR36 at indicated age. Note that PR36 caused motor neuron loss when compared to the control PA36, and the neuronal loss progressed with aging.
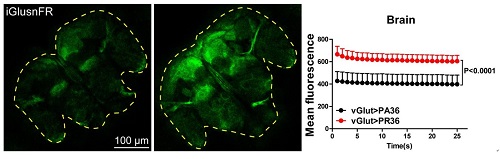
Fig. 2. Representative images showing the increased extracellular glutamate signals detected with sensor iGluSnFR in adult brain of arginine-rich DPR expressing flies.
 附件下载:
附件下载: 
