Hereditary neural disease refers to a disease with the main clinical manifestations of nervous system dysfunction in developing individuals due to changes in the genetic material of germ cells or fertilized eggs. It includes chromosomal aberrations, polygenic and monogenetic diseases, accounting for around 50% of monogenetic diseases. Hereditary neural disease has congenital and lifelong characteristics with high disability and mortality, which affects patients for life. To this day, the etiology and pathogenesis of many hereditary neural diseases have not yet been elucidated, lacking effective clinical diagnosis and treatments. Elucidating the molecular basis of its pathogenesis will promote basic research and clinical application of neural genetics. Our research group has visited hundreds of villages in more than 10 provinces since 2004, and has collected and characterized about 220 families with genetic diseases, 70% of which are hereditary neural diseases. By molecular genetic analysis, we found 138 mutations involving 45 disease-causing genes. These results play an important role in accurate clinical diagnosis and prenatal diagnosis of these hereditary neural diseases. We were the first to identify mutations in SLC20A2, SCN11A, and SN genes associated with idiopathic basal ganglia calcification (IBGC), episodic pain, and essential tremor (ET) diseases, respectively. At present, we mainly focus on the molecular pathological mechanisms of IBGC, episodic pain, and ET disease, and the development of targeted prevention and treatment drugs in further research.
Pathogenesis of IBGC disease and drug development for prevention and treatment
IBGC, also known as Fahr disease, is characterized by abnormal calcium deposition in the basal ganglia and other brain regions and a wide spectrum of neuropsychiatric symptoms including Parkinsonism, dystonia, tremor, ataxia, dementia, psychosis, seizures and chronic headache, leading to patient suffering. Calcification of the basal ganglia is observed in 1-2% of CT scans. In 1850, IBGC disease was first described by Delacour, which commonly showed autosomal dominant and recessive inheritance. Until 2012, the pathogenesis of IBGCs remained unknown. We were the first to identify mutations of SLC20A2 in IBGC patients and found that the SLC20A2 mutations resulted in a significantly impaired phosphate transport. This disease was associated with brain calcification and local phosphorus homeostasis for the first time. The result was published in Nature Genetics in 2012. Since then, researchers have identified many mutations of this gene in IBGC patients. The mutation rate of SLC20A2 in IBGC patients is 40-50%.
How does the SLC20A2 mutation cause IBGC? The SLC20A2 encodes type III sodium phosphate transporter 2 (PiT2), which consists of 652 amino acids including 12 transmembrane regions, 5 extracellular regions and 6 cytoplasmic regions according to bioinformatics analysis. The 7th cytoplasmic region is composed of 246 amino acids (also known as loop7, accounting for 38% of the amino acid composition of the whole protein) between two PD domains (highly conserved amino acid sequences throughout evolution) of PiT2. The PD domains of PiT2 are related to Pi transport. PiT2 is an electrical sodium phosphate transporter. What is the actual mechanism underlying how PiT2 mutants cause Pi transport deficiency in human IBGC patients? What is the consequence of the mutation in loop7 of PiT2? How to prevent and treat IBGC disease?
In order to answer these questions, we will conduct investigations in the following 4 aspects: (1) We will research the effects of PiT2 mutations on Pi transfer function and electrophysiological charaterization to reveal the molecular pathological mechanism underlying IBGC caused by SLC20A2 mutation; (2) We will study how loop7 domain regulates the membrane localization of PiT2 by yeast two hybridization and other molecular biology strategies; (3) We have constructed the mouse model with Slc20a2 mutation and will reveal how SLC20A2 mutation leads to IBGC in vivo, which will serve as a critical foundation for subsequent drug development; (4) We will develop drugs for prevention and treatment of IBGC. We have discovered that the LJY-001 Chinese medicine complex inhibits intracranial calcification in mice with Slc20a2 mutation. Next, we will answer the following questions: What is the mechanism underlying the effects of LJY-001? Is it a PiT2 target drug? Does it work on other ectopic calcifications? What are the main active ingredients in the LJY-001 complex? What is the toxicity of LJY-001 and so on.
Pathogenic mechanism of episodic pain and drug development
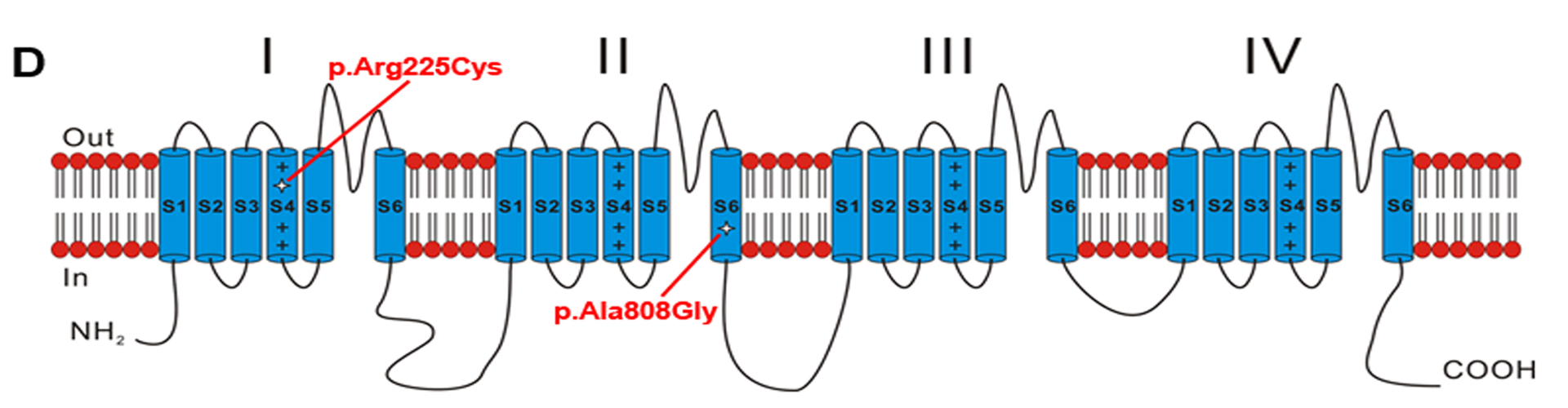
Pain serves as a defense system to protect the body against further injury and promotes healing of damaged tissues. Nociceptors detect noxious stimuli and produce the sensation of pain, and such a pain mediating signal is conveyed to the central nervous system (CNS) by means of action potentials. The voltage-gated sodium ion channels (VGSCs) are essential for the generation of action potentials in excitable cells. VGSCs consist of pore-forming α subunits that encode the core protein of the channel and associated β subunits that modify the channel function. Ten α subunits of VGSCs (Nav1.1–Nav1.9 and an atypical channel Nax) have been identified in mammals and are encoded by SCN11A–SCN5A, SCN8A–SCN11A, and SCN7A, respectively. Nav1.7, Nav1.8, and Nav1.9 are preferentially expressed in peripheral somatosensory neurons and have been implicated in injury-induced neuronal hyperexcitability. Nav1.7 and Nav1.8 have been linked to human hereditary pain. Yet, as of 2013, a direct link of Nav1.9 to human pain has not been reported.
Our team reported two large Chinese families including 28 patients with autosomal dominant episodic pain and found two missense mutations in SCN11A gene encoding voltage-gated sodium channel Nav1.9 (SCN11A), c.673C>T (p.Arg225Cys) and c.2423C>G (p.Ala808Gly) (one in each family). Both mutations enhanced the channel’s electrical activities and induced hyperexcitablity of DRG neurons. We discovered for the first time that SCN11A gain-of-function mutations caused human pain, and the result was published in Am J Hum Genet in November of 2013. At the same time, two patients with sporadic painlessness carrying gain-of-function SCN11A mutations was reported in Nat Genet. Does the gain-of-function Nav1.9 mutation cause pain or painlessness? What are the detailed pathological mechanisms of pain disorders caused by Nav1.9 mutations? We will investigate the following three aspects: (1) The relationship between genotype and phenotype of SCN11A mutations leading to pain disorders; (2) The effects of known SCN11A mutations on the function of Nav1.9 channel; (3) SCN11A knock-in and knock-out mouse models will be constructed to study the molecular consequences of Nav1.9 mutations in pain disorders and develop analgesic drugs targeting Nav1.9.
Studying the function of novel pathogenic gene of essential tremor
Essential tremor (ET) is one of the most prevalent neurological disorders characterized by hyperkinetic tremor, which worsens with movement. The estimated prevalence of ET in the general population is 1%, which increases with age and accounts for 5% of the individuals >65 years old. ET is typically characterized by involuntary, rhythmic shaking of one or more parts of the body (including upper extremities, head, voice, tongue and chin etc.), and there are both kinetic tremor (voluntary movements) and postural tremor (positions against gravity). ET patients are 24 times more likely to develop Parkinson's disease in later life than the normal population, which means ET may be an important risk factor for Parkinson. In clinical practice, there are no obvious biomarkers in ET patients and no clear etiology, which has been puzzling clinicians in terms of the precise diagnosis and targeted treatment of ET patients. The identification of large ET families as well as twin studies indicate that genetic factor has an important role in ET etiology. However, the lack of diagnostic biomarkers leads to high misdiagnosis rate and there is incomplete penetrance in familial and sporadic ET cases, therefore it is hard to find the disease-causing gene of ET.
Our group collected and charcterized 144 Chinese families with ET, and have identified a candidate disease-causing gene SN by exome sequencing. The mutation rate of SN gene in ET patients is 7.6%. However, the function of the SN is unknown. After immunofluorescence analysis, we found that this gene is mainly expressed in Purkinje neurons in the cerebellum. We have constructed sn knockout ??mice and conditional knockout mice of Purkinje neurons, and found that loss-of-function mutation in sn resulted in tremor phenotype. After further investigation, we found structural changes in Purkinje neurons of the cerebellum in sn-KO and coditional KO mice.
In the next steps, we will mainly focus on the molecular pathological mechanism of ET caused by SN mutation from the following three aspects: (1) The relationship between the genotype and phenotype of SN mutations; (2) What is the main function of the SN protein? How does the known SN mutation affect the function of SN protein? How does SN mutation affect the feed-forward/feedback homeostasis of the cortical-olivo-cerebellar-thalamic neural circuit? We will analyze the role of neural circuits containing PC neurons and other neurons in the cerebellum in the pathological mechanism of ET; (3) Can wild-type SN gene, SN substrate, and activators/inhibitors of key molecules in PC neurons identified by RNA-seq rescue the phenotype of SN-KO mice?

Senior Investigator